The Research Institute for Computational Science at the National Institute of Advanced Industrial Science and Technology (AIST) has been successful for the first time in analyzing in-depth the catalytic reactions of ribozymes, which have a promising potential as a gene therapy drug for diseases such as cancer. The study was carried out by using a computer simulation technique involving the first-principles quantum mechanics-based and molecular dynamics calculation method. This technique holds great promise of contributing to the development and design of novel and more effective gene therapy.
The conventional view has been that all “enzymes” are “proteins.” In the latter half of the 20th century, however, it was discovered that there are some ribonucleic acids (RNA) which also act as enzymes. These enzymes are called ribozymes. Because they act as “scissors” cutting themselves off from other RNAs, they have a significant potential for application in gene therapy involving the cutting-off of the causal gene responsible for the disease such as cancer.
Although it is known that the catalytic reactions of ribozymes require the presence of metal ions as catalysts, the mechanism for the reaction is not fully understood. We have used the First-Principles Molecular Dynamics Calculation Method, a technique combining quantum mechanics (First-Principles) describing the chemical reactions and the molecular dynamics calculation method tracing the thermal oscillations of the molecules, to reproduce the ribozyme catalytic reaction processes in the computer. With this technique, we were successful for the first time in studying the reaction pathways and in-depth mechanism which cannot be traced by experimental methods.
Although the ribozyme reaction pathways are diverse and extremely complex, organisms are capable of correctly selecting only those reactions that are desirable. The results of this simulation-based analysis have demonstrated for the first time that the metal ions bonding to the ribozyme molecules not only have the already-known catalytic action but also have a pathway-marshaling function to select and extract only the correct reaction pathways.
The computation results are in good agreement with the conventional experimental findings, and their significance is that it has become possible to predict with this computational technique which reaction pathways the molecular system will (or will not) go through. The further development of this computational technique has therefore a very promising potential for opening up new avenues of developing and designing, with use of computers, more effective therapeutic procedures (drugs) in the gene therapy domain.
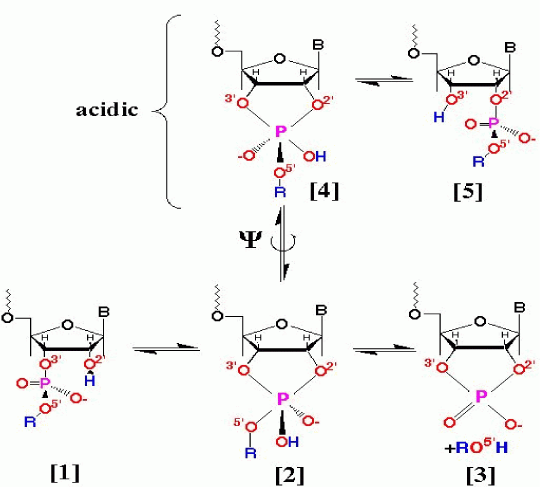 |
|
Fig. 1. In the ribozyme reaction, several paths are competing (part of the reaction paths are indicated above); thus the reaction mechanism is complicated. When the reaction proceeds along [1] to [3], the specific site is hydrolyzed. Therefore, this reaction path can be used for cancer gene therapy by inhibiting the expression of oncogene. |
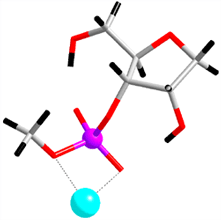 |
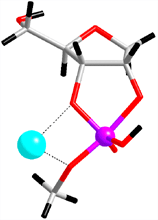 |
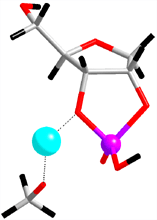 |
|
(i) |
(ii) |
(iii) |
 |
|
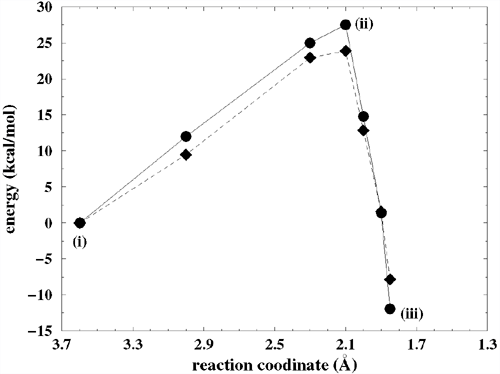
Fig. 2. Detailed mechanism of the catalytic reaction of a ribozyme, revealed by the first-principles molecular dynamics simulation. Upper: (i) the initial structure of the simulation, (ii) the transition state, (iii) products. Below: the free energy (indicated by diamonds) and enthalpy (balls) profiles throughout the reaction process, calculated from the simulation. |