量子物理×深層学習でAIがおおきく進化
量子物理×深層学習でAIがおおきく進化

2022/08/31
量子物理×深層学習でAIがおおきく進化 学習対象外の領域でも高精度に予測

 研究室では高い性能を示した人工知能(AI)が、実際の現場では思ったように動かないというのはよく聞く話だ。この最大の原因は、深層学習技術で開発したAIは「融通が利かない」こと。学習に使ったデータの範囲を少しでも外れると、AIが途端に役に立たなくなってしまう。
研究室では高い性能を示した人工知能(AI)が、実際の現場では思ったように動かないというのはよく聞く話だ。この最大の原因は、深層学習技術で開発したAIは「融通が利かない」こと。学習に使ったデータの範囲を少しでも外れると、AIが途端に役に立たなくなってしまう。
AIが次の段階に進むために、産総研は、学習データから外れる「外挿領域」でも適切に予測ができる新たな深層学習の基礎技術を開発した。多くの競合研究がある中で、この技術の他とは一線を画す優れた特徴は、学習に使ったデータから外れた領域でも適切に予測ができること。分子サイズが小さい物体の特性データで学習させただけで、分子サイズが大きい物体の特性も精度よく見積もれるというわけだ。実現のカギは、深層学習と量子物理学の知見の融合である。学習データの外で高い性能が出せないことは、従来の深層学習技術の大きな弱点であった。この弱点を深層学習と理論の融合で越える今回の技術には、材料開発、創薬をはじめとした物理・化学に関連する分野でまったく新たな成果を生み出すことが期待できそうだ。
最高評価のAIが現場で使えない
発端は苦い経験だった。人工知能研究センターの椿 真史は、薬剤とタンパク質の相互作用を予測する深層学習技術の研究をしていた。2017年12月、研究の成果をAI分野で最高峰といわれる国際会議で報告し、参加したワークショップのベストペーパー賞を見事に受賞する。ところが、この技術を製薬企業と協力して現場で活用しようとしたところ、有望な結果が全く得られなかったのだ。
実際のところ、実験では高い性能を示したAIが、現場では思ったように動かないことはよくある話だ。最大の問題は、深層学習技術で開発したAIは融通が利かないことである。学習に使ったデータの範囲を少しでも外れると、AIは途端に役に立たなくなってしまう。例えば晴天や雨天だけのデータで学習させた自動運転車は、雪が降るとお手上げとなる。「黒い猫」や「白い犬」の写真で学んだ画像認識AIは、そこに映った動物が「“白い”猫」や「“黒い”犬」の場合、適切な判断が下せない。
この問題を解決するために、世界中で研究が進んでいる。欧米の大手IT企業はその潤沢な資金力を武器に途方もなく大量のデータを集めて、最新鋭のスパコンでも学習に何日もかかるほど巨大なAIモデルを開発している。力技で、大量のデータを集めてまったく見たことのないようなデータを限りなく減らす手法といえる。
椿たちのチームは、もっと効率的な方法はないだろうかと物理法則の理論に目をつけた。物質の性質を考える基礎となる量子物理学の理論と、大量のデータに頼る深層学習を適所で組み合わせる方法である。(2020/11/11プレスリリース記事)
初めてのデータ領域でも高精度に
チームが実現したのは、物体の分子の構造(分子を構成する原子の位置関係)のデータを入力すると、分子の特性(エネルギー)を予測できるAIである。このAIの学習には、さまざまな物体の分子の特性を調べた既存のデータベースを用いた。
AIの予測能力を調べるために、椿らはあらかじめこのデータベースを学習用のデータとテスト用のデータに切り分けた。ポイントは、学習用データが分子サイズの小さい(原子の数が少ない)物質だけを含むのに対し、テスト用データは分子サイズが小さい物質と分子が大きい(原子の数が多い)物質の両方を含んでいることだ。前者でAIを学習させて、後者を使って、分子サイズの小さい物質から大きい物質までさまざまな分子の特性を予測させた。
その結果が次の図である。横軸が分子サイズの大きさ(原子の数)、縦軸は正解と比べた誤差の大きさを表す。赤い線が椿らの開発したAI、青い線が従来技術で、図中の「外挿領域」が学習用のデータから外れた領域のことである。
従来技術との差は歴然だ。新開発のAIは、初めて接するデータの領域において、予測の誤差を大幅に減らすことに成功した。
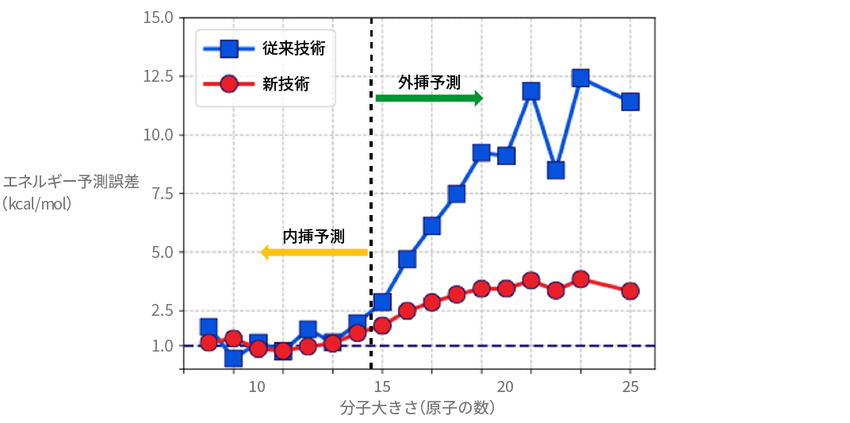 物性値(エネルギー)の外挿予測精度
物性値(エネルギー)の外挿予測精度
「外挿領域」における誤差を大幅に削減したことがわかる
新開発のAIが1万種類の分子特性を数分で予測
新材料の開発の第一歩は、候補となる分子の特性を見積もることである。世の中にあり得る分子の構成(原子の組み合わせ方)は数え切れず、特性がわかっているのはそのごく一部にすぎない。AIによって分子の特性を予測できれば、多大な時間や費用がかかる実験をせずとも、あらかじめ新材料の候補を絞り込めることができる。
これまでにも、実験をせずに分子の特性を調べる手段はあった。物理法則に基づいて、分子の状態をコンピューターでシミュレーションする方法である。ところがこの方法では、簡単な有機化合物を1種類調べるだけで数十分から数時間もかかってしまう。
椿らが開発したAIは、計算にかかる時間を一挙に短縮。1万種類もの分子の特性を、わずか数分で予測することができるのだ。コンピューターシミュレーションに比べ、実に10万倍以上も高速である。
しかも、両者の差は物質の大きな分子になるほど拡大する。コンピューターシミュレーションでは、分子が含む電子の数が増えると、計算時間が電子数の3~4乗の割合で長くなる。例えば電子数が2倍になると、計算時間は2の3乗~4乗、つまり8倍から16倍にも増えてしまう。
このAIであれば、分子のサイズが大きくなっても短時間で結果を出せ、しかも、小さな分子のデータを学習するだけで、大きな分子の特性を精度よく予測できる。これまで得られなかった情報を短時間で探り出し、材料開発に大きな変革をもたらす可能性がある技術なのだ。

量子物理学の知見を導入
では、新技術はどうやって既存の技術の欠点を補ったのだろうか。
これまでの深層学習技術では、AIが分子構造のデータと、その特性のデータのペアを大量に見比べて、構造と特性の間に成立する「公式」を求めてきた。ニューラルネットワークを用いたAIは複雑かつ巨大な数式と言い換えることもでき、この数式の係数(パラメータ)を適切に調整したうえで、分子構造データを入力すると、特性のデータを出力する公式を導いてくれる。別の言葉でいうと、深層学習は途中経過を気にせず、結果の数字が合っていることを重視した方法といえ、学習データの範囲では的確な結果であっても、学習データの範囲から一歩でも外れると、AIは見当違いの結果を出す公式を学びかねないということになる。
そこで椿らは、入力と出力の間の途中経過に存在する理論的な関係と深層学習を、「いいとこ取り」で組み合わせることにした。理論的な関係との組み合わせとは、分子特性を調べるシミュレーションでも利用している量子物理学の知見に基づいて組み立てる。まず、物理の知見から適切に素早く計算できる部分はAIの学習対象から外し、深層学習による「数字合わせ」の部分を最小限に止めることで、幅広い範囲で通用する公式を求めようとしたのである。
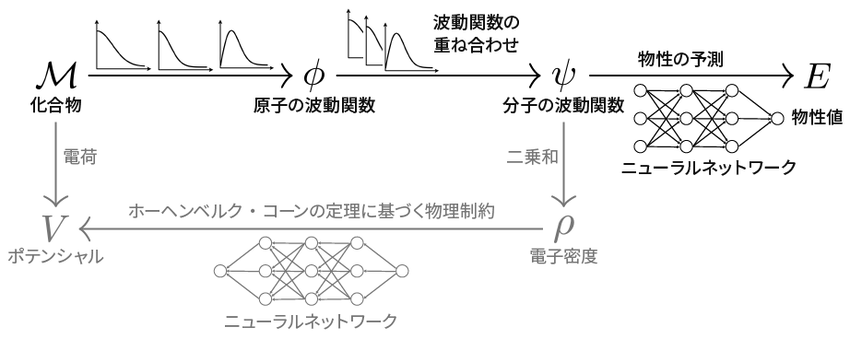 今回開発した深層学習モデルの概略図
今回開発した深層学習モデルの概略図
まずは左端の化合物Mから右端の物性値Eの流れに注目
上の図を見てほしい。左上にある「化合物M」が分子の構造、右上にある「物性値E」が分子の特性に当たる。これまでの深層学習は、MとEのデータだけを見て、両者をつなぐ公式を作っていたことになる。
ただし物理学的にMからEを導くには、原子の状態(原子の波動関数φ)や分子の状態(電子の波動関数ψ)をまず知ることが必要である。椿らはこの部分に、量子物理学で知られる公式を導入した。一方で、分子の波動関数ψから物性値Eを予測する部分は計算が非常に複雑なので、ここは深層学習によるAI(ニューラルネットワーク)に任せる。あとは、物理学の公式の係数とAIの内部の係数を、データから一気に学習するのである(図のM→φ→ψ→E)。
途中の結果でも答え合わせ
もっとも、物理学の公式とAIを組み合わせただけでは、まだ十分とはいえない。両方の係数を学習した結果、それでも狭い範囲でしか通用しない公式になってしまう可能性は残るからである。そこで椿らは、物理学の公式が正しい形に落ち着くように、もう一つの制約を加えることにした。それが図の下半分である。
量子物理学の知見から、化合物の分子中における電子の密度(ρ)と、分子の電気的特性(ポテンシャルV)の間には、一定の関係が成り立つ(ホーンベルク・コーンの定理)。両者の間の公式は、深層学習によるAI(ニューラルネットワーク)で表現可能である。しかも、分子の密度ρは、波動関数ψから簡単な数式(二乗和)で求められる。つまり、化合物MからポテンシャルVを予測する公式(図のM→φ→ψ→ρ→V)を作ることができるわけだ。
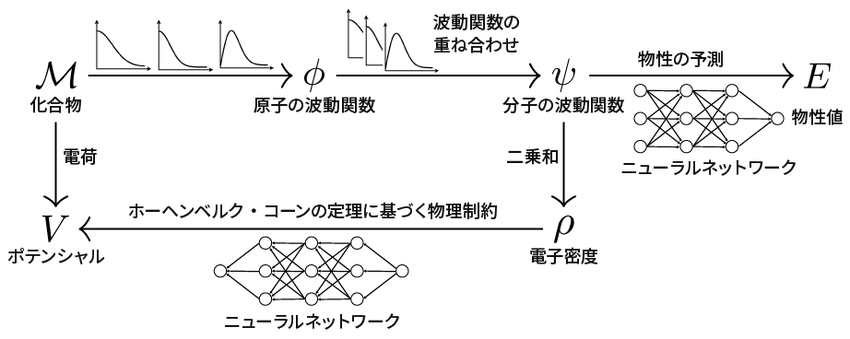 量子物理学の知見から制約を加えて、深層学習モデルの精度を上げる
量子物理学の知見から制約を加えて、深層学習モデルの精度を上げる
ここで、化合物MごとにポテンシャルVがどれくらいになるかのデータをたくさん使えば、MからVの公式の係数を学習することが可能となる。これによって、化合物Mから分子の波動関数ψを求める公式が、物理学的に正しい方向に近づく。あとは、この制約の経路の学習と、先ほどの物性値を求める経路の学習を交互に繰り返せば、図中にある全ての公式がどんどん正解に近づいていく手法だ。
この方法で開発したAIが、物性値を正しく予測できることは前述した。もう一つの「答え合わせ」は、制約に用いた電子の密度ρを正しく推測しているかどうかである。その結果が次の図だ。黄色の点線がシミュレーション結果、赤い線がAIの出力である。AIの出力は、二つのピークがあるといった、シミュレーション結果の特徴をうまく捉えている。
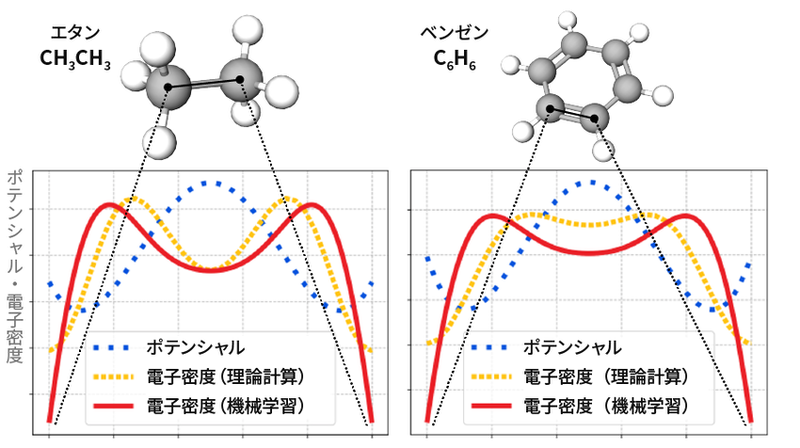 エタンとベンゼンの化学結合の電子密度の比較
エタンとベンゼンの化学結合の電子密度の比較
シミュレーションで得られる電子密度(黄色)には二つのピークがあり、今回開発した深層学習モデル(赤)でもその二つのピークを再現できた。
5年で分子特性、10年で化学反応の仕組みを解明
この技術には、まだまだ改良の余地が残されている。電子の密度や物性値の予測結果を見ると、どちらもそれなりの誤差があり、より実用的なモデルにするにはこの誤差を減らすのが一つの目標になる。材料の分子の中には、多くの原子が3次元的に複雑な配置で結合したものがあり、そうした構造の情報をAIの計算に含めるといった改善方法が考えられるという。
さらに難易度が高い目標としては、AIが予測する分子の特性を、現状のエネルギーの他にも広げることだ。コンピューターシミュレーションでは、エネルギーを求める過程の副産物としてさまざまな特性が得られる。ところが深層学習を活用したAIからわかるのは最終的な結果だけで、途中の経過が何を表しているのか不明な場合がほとんどである。現在の深層学習の学習モデルが、中身の見えない「ブラックボックス」と呼ばれるのはこのためだ。
椿らは、今回のAIが学習した後のモデル内のパラメータ(数値)に、エネルギー以外の様々な物理情報が埋め込まれていると見ている。この学習済みモデル内のパラメータを基にして、さまざまな特性を予測する手法の研究を進めているところだ。
分子ごとの特性の先には、開発した触媒が化学反応をどれだけ促進するのか、薬剤の候補がタンパク質とどのように結合するのかといった、より高度な現象を予測するAIの研究が控えている。「どのくらいの時間がかかりそうですか」と聞くと、椿はこう答えた。「5年後までに物理、10年後までに化学、生物はその先ですね」。物理とは、ある材料の分子の特性をつまびらかにすること。化学とは、複数の分子が関わる化学反応の仕組みの解明を指す。そして生物の段階では、タンパク質をはじめとする極めて複雑な分子の相互作用が対象となる。
これだけ幅の広い課題は、椿ら小規模の研究グループだけで解決できるものではない。椿は産業界やアカデミアから、協力してくれる研究者を募っている。触媒や薬剤といった応用分野の専門家はもちろん、言語など異なる対象を扱うAI研究者の視点も取り入れたいという。
AIを用いた材料開発は、世界中の企業や研究機関が激しい競争を展開する激戦区である。この分野で独自の手法により大きな成果を上げれば、日本の産業に大きな恩恵をもたらすだけでなく世界にインパクトを与えるだろう。
椿は自分たちが生み出した基礎研究の種を、産官学の協力で芽吹かせ、大きく育て、日本企業の材料開発や創薬に貢献するのみならず、物理学・化学に関連する多くの分野に変革を起こす日を目指して今日も研究を進めている。
情報・人間工学領域
人工知能研究センター
機械学習研究チーム
研究員
椿 真史
Tsubaki Masashi