国立研究開発法人 産業技術総合研究所【理事長 中鉢 良治】(以下「産総研」という)人工知能研究センター【研究センター長 辻井 潤一】機械学習研究チーム 瀬々 潤 研究チーム長、椿 真史 産総研特別研究員は、インテリジェントバイオインフォマティクス研究チーム 富井 健太郎 研究チーム長と共同で、2種類の深層ニューラルネットを組み合わせて薬剤とタンパク質の相互作用を予測する手法を開発し、大規模データを用いた実証実験により高速で高精度に予測および相互作用部位候補の特定ができることを示した。
近年、創薬分野への人工知能技術の応用による新薬開発の加速化や、革新的な新薬の実現が期待されている。しかし薬剤とその対象となるタンパク質は異なるタイプの構造なので、疾患治療に有効な薬剤とタンパク質の組み合わせを高速で高精度に予測することは困難であった。そこで、化学と生物学の知識に基づき、性質の異なる深層学習手法を組み合わせて、薬剤とタンパク質に対する高速で高精度の相互作用予測手法を開発した。また、薬剤やタンパク質の立体構造を用いない従来手法では、予測結果から相互作用部位を特定できず、結果の解釈に問題があるとされてきたが、今回開発した手法では相互作用部位を特定、可視化できる。この技術により、新薬剤を大量の候補群から絞り込む作業の加速が期待される。さらに、コンピューターによる大規模な薬剤候補の探索によって、人間の知識や経験だけでは到達できない革新的な新薬の開発も期待される。なお、今回開発した手法の詳細は、2018年7月6日に学術雑誌Bioinformaticsに掲載された。
 |
|
今回の手法で予測した薬剤とタンパク質の相互作用部位の可視化例 |
近年、機械学習技術の一つとして大きな成功を収めている深層学習を、創薬をはじめとする化学・生物学分野へ応用することが期待されている。例えば、薬剤とタンパク質の相互作用が、深層学習によって高速で高精度に予測できれば、新薬開発を加速させるだけではなく、人間の知識や経験だけでは到達できない革新的な薬剤の開発が期待される(例えばNature 557, S55-S57 (2018); doi: 10.1038/d41586-018-05267-x)。しかし、薬剤とタンパク質は、それぞれ異なるタイプの構造であるため、双方のデータを深層学習でどのように統一的に扱うかが大きな問題となっていた。さらに深層学習は予測結果の解釈が難しく、化学・生物学分野への応用の障壁となっていた。
産総研は、機械学習を、画像や言語などのデータだけでなく、化学・生物学のデータへも応用することを目指している。特に近年は、深層学習技術の創薬応用に取り組んできた。
なお、本研究開発は、国立研究開発法人新エネルギー・産業技術総合開発機構(NEDO)「次世代人工知能・ロボット中核技術開発/次世代人工知能技術分野/人間と相互理解できる次世代人工知能技術の研究開発」、独立行政法人日本学術振興会(JSPS) 科研費JP15H01717、JP17H07392、国立研究開発法人日本医療研究開発機構(AMED)創薬等ライフサイエンス研究支援基盤事業創薬等先端技術支援基盤プラットフォーム(BINDS)「タンパク質の高次構造情報を利用した創薬等研究加速に向けたバイオインフォマティクス研究」、国立研究開発法人科学技術振興機構(JST)、CREST、JPMJCR1689の支援を受けて行った。
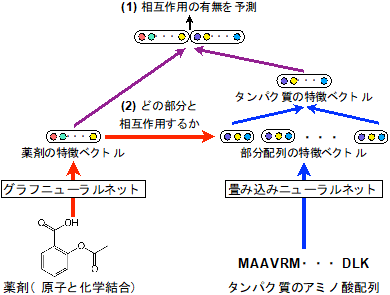
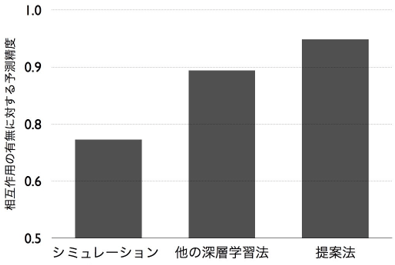
薬剤は原子と結合をグラフ構造データとして、タンパク質はアミノ酸の配列を配列構造データとして、それぞれ表現できる。しかし、これまでこれらの異なる2種類のデータに共通して有効な深層学習手法がなかった。今回開発した手法では、各薬剤のデータに適した深層学習手法であるグラフニューラルネットと、タンパク質のデータに適した深層学習手法である畳み込みニューラルネットをそれぞれのデータに適用して、薬剤とタンパク質それぞれの性質を適切に捉える特徴ベクトルを計算する。薬剤とタンパク質の大規模なデータを用いてこの特徴ベクトルを学習することで、相互作用の有無を予測する(図1)。3万5千以上の薬剤とタンパク質の相互作用のデータを用いた実証評価実験により、従来用いられていたものよりも低次元(10次元程度)の特徴ベクトルを用いても相互作用の有無を適切に予測できることが分かった。低次元の特徴ベクトルを用いるため計算量が抑えられ、高速な予測が実現できた。また精度も、既存のドッキングシミュレーションや近年開発された他の深層学習手法に比べて、高い予測精度(既存手法比で3%~10%の向上)を示した(図2)。また、それらシミュレーションや他の深層学習手法は、薬剤やタンパク質の立体構造データを入力することが必要で、立体構造データ自体が入手しにくい上に、大規模計算になってしまい、計算コストが高い。一方で今回の手法は、薬剤のグラフとタンパク質の配列の情報のみから予測できるため、立体構造がまだわかっていない膨大な数のタンパク質についても適用可能である。
 |
|
図1:今回開発した薬剤とタンパク質の相互作用の予測手法 |
 |
|
図2:既存の手法と今回開発した手法の予測精度の比較 |
深層学習は高速で高精度な予測ができる一方で、予測結果の解釈が難しい。化学・生物学のデータを扱う上では、コンピューターによる自動的な予測結果に対し、人間が既に持っている化学・生物学の知識と照らし合わせて、結果の妥当性を判断する必要がある。そのため、結果を解釈しやすい機械学習技術は重要である。そこで、相互作用の有無という大まかな予測(図1中の(1))に加えて、2つの特徴ベクトルを用いて、薬剤が相互作用しやすいタンパク質の部位という、より細かい情報(図1中の(2))も考慮した予測を行い、その結果を可視化した。予測した相互作用部位を可視化することによって、化学・生物学の知識と照らし合わせての検証が容易にできるようになった。
今後は、薬剤やタンパク質の三次元立体構造をより詳細に考慮した手法を開発し、さまざまな薬剤とタンパク質を用いて、その相互作用部位の立体構造を網羅的に検証し、予測結果の信頼性を高めていき、新薬開発支援による創薬分野への貢献を目指す。